

���X��(gu��)�Ҍ�(sh��)�(y��n)�ң��o����O���ϵ��Ƃ估����
�r(sh��)�g:2020-10-16 21:37��Դ:�����Դ ����:����
�c(di��n)��:
��
������B
��x��늳����Ƅ�(d��ng)��(sh��)�֕r(sh��)�����g(sh��)��������Ҫ�M�ɲ��֡���������S��늄�(d��ng)��܇�ij��F(xi��n)����(d��ng)ǰ늳��о�����Ҫ���c(di��n)���D(zhu��n)��ͳɱ�늳ص��_�l(f��)��Ŀǰ��늳ز����л����������Ԫ�ء����磬�������LiNixMnyCozO2(NMC)��LiNi0.8Co0.15Al0.05O2(NCA)���O���ϡ�⒵��_�ɺ;����Č�(du��)늳��ИI(y��)�ĵͳɱ��Ϳɳ��m(x��)Ŀ��(bi��o)��һ��(g��)����(zh��n)������ȫ��⒃�(ch��)����Ѹ�ٜp��Ҳ�ӄ���Ⓓ���(y��ng)�����ƣ��@һ���ƌ���u�_ʼ���{늄�(d��ng)��܇�Ј�(ch��ng)��δ�������⣬������������ӣ�⒵ăr(ji��)�����^ȥ��������L(zh��ng)�˃ɱ�������_�l(f��)Ⓔ����^�͵����O�������P(gu��n)��Ҫ����ˣ�Ŀǰ�����(sh��)늳��ИI(y��)ʹ�õ�NMC��NCA���O�������nj�(sh��)�F(xi��n)�@ЩĿ��(bi��o)������x���-�������(LiNiO2)�mȻ��һ��(g��)�аl(f��)�����ǣ�LiNiO2�^�y�ϳɣ���늻��W(xu��)�y(c��)ԇ�^���нY(ji��)��(g��u)��(hu��)�l(f��)����(y��n)�ص�׃������(d��o)��ѭ�h(hu��n)�����^����i������(LNMO)��4.9 V�⾧ʯ���O���ϣ�����һ���u�̘I(y��)���ĺ��x���ϡ�Ȼ�����@�N���ϵ������^��(��Փ�ϼs��147 mA h/g)����Ҫ���^��늉����Ҍ���늽�Һ�wϵ�²������x��Ȼ������(d��ng)���^��늉�ѭ�h(hu��n)�r(sh��)���@Щ���ϵ�����˥�p�^�졣
�ɹ���(ji��n)��
��������X��(gu��)�Ҍ�(sh��)�(y��n)�ҵ�Nitin Muralidharan(ͨӍ����)��Ilias Belharouak(ͨӍ����)��(b��o)����һ�N�ܵ��͵IJ��ϣ������F����NCA�Y(ji��)��(g��u)�е�⒣��Ķ��γ�һ�NⒺ�����“��”���������O���ϡ��@�N�µğo����O���ϣ�ͨʽ��L(zh��ng)iNixFeyAlzO2(NFA)���ڲ��Ɖ�NCA�Y(ji��)��(g��u)�Ͱ�ȫ��(y��u)��(sh��)����r������⒣�ͬ�r(sh��)�����������懺���(>80%)��������������������������r(ji��)�X���F���沿��懣����r(ji��)�X���F���x�Ӱ돽�cNi3+����(Al3+�x�Ӱ돽��0.54 Å��F(xi��n)e3+��0.55 Å����Ni3+���x�Ӱ돽��0.55 Å)�������ڽY(ji��)��(g��u)��(w��n)���Եĸ��ƣ�����(qi��ng)��ȫ�ԣ�������ѭ�h(hu��n)���������Ĉ�(b��o)����NFA���O������ͨ�^���������ϳɵģ��������õı������ܺͷ�(w��n)����ѭ�h(hu��n)���ܣ�ͬ�r(sh��)�����^�ߵ����������P(gu��n)�о��ɹ���LiNixFeyAlzO2, a new cobalt-free layered cathode material for advanced Li-ion batteries���}�l(f��)����Journal of Power Sources�ڿ��ϡ�
�D�Ľ���
������260Ӣ���늄�(d��ng)��܇��Ҫ76.47 kW h��늳ؽM�������c�o�NFA��ȣ����NNMC���O��������������D1(A)��ʾ����(du��)��ʹ��NFA�������O���ϣ���ͬ��(sh��)���ľ�����ͬ��̺�늳ؽM������늄�(d��ng)��܇������������“0”���@�o��ͻ���˟o����O���ϣ���NFA��(j��)��������һ��늳�ϵ�y(t��ng)�ĝ������D1(B)������(g��)�^�ɽ��ٵ�pHֵ�S�A��Һ(NaOH)�w�e�����Ӷ�׃���ij���څ��(sh��)������څ��(sh��)���Ը���(j��)(��1)�е��ܽ�ȷeKsp����ጡ����Կ�����Ni2+→Ni(OH)2��Fe2+→Fe(OH)2��Kspֵ����Al3+→Al(OH)3��ͬ�r(sh��)����(j��)���ߵõ��ij���څ��(sh��)�������Ɣ�����c�������(pH~6)�͚������F(pH~8)��ȣ��������X�ڷdz��͵�pHֵ(~2)�r(sh��)�����_ʼ���������⣬���^��pHֵ(>12)�£������Ě������X��(hu��)��ȫ�ܽ⡣�@��ζ��������Al(OH)3��Ksp��Ni(OH)2��Fe(OH)2С�ö࣬����Ni2+��Fe2+��Fe3+��Al3+��ͬ�r(sh��)��������һ��(g��)����(zh��n)����һ���棬懺��F�Ě���������pHֵ>6�r(sh��)�_ʼ�������@�M(j��n)һ���sС��ͬ�r(sh��)���������ڡ����]���@Щ���أ�NFA���Ϗĺ��(80%)���F(10%)���X(10%)�Ļ���^�ɽ����}��Һ�й��������c��(g��)Ԫ�صij���څ��(sh��)��ȣ�NFA����څ��(sh��)���������pHֵ>10��<12���t���Ԍ�(sh��)�F(xi��n)���NԪ�ص�ͬ�r(sh��)�������������@Щ�Y(ji��)���������A(y��)Ӌ(j��)�㶨��pHֵ~11.5���Դ��M(j��n)NFA���������D1(C)���Ƃ�����NFA(OH)2ǰ�(q��)�w���џ��ԫ@��LiNFAO2���O��ĩ������(g��)NFA���O���Ϻϳ��^�̵�ʾ��D�����ڹ��������о���������������4 L��pH�㶨��11.5���B�m(x��)����۷���(y��ng)��(CSTR)���M(j��n)���˹������^�̡�CSTR����(y��ng)��ʹ�����^�ɽ��ٵ���Һ(Ni��Fe��Al)�c�A��Һ(NH4OH��NaOH)���ܿط�ʽ����(y��ng)���B�m(x��)���ɚ�������ǰ�(q��)�w(NFA(OH)2)�����������(y��ng)����(n��i)��~4 Lȥ�x��ˮ�M�ɣ�ԓȥ�x��ˮ��500 rpm�²������裬�γ���ҺA������(y��ng)����(n��i)�ij�ʼpH�ӽ����ԣ������һ�����ӉAҺ(��ҺC)��ԓ��Һ���m��(d��ng)Ħ����ȵ�NH4OH��NaOH�M�ɣ��˕r(sh��)����(y��ng)����(n��i)��Һ��pHֵ��ߵ�~11.5��������pH��ֵ���Ա�ֻ�Ю�(d��ng)����(y��ng)����(n��i)�����pHֵ��11.45��11.55֮�g�r(sh��)���ŕ�(hu��)�����^�ɽ�����Һ(��ҺB)���ķ���(y��ng)�����B�m(x��)�ռ��������a(ch��n)��ռ�ֱ���@�ú��о��������ΑB(t��i)��ǰ�(q��)�w���ӵķ�(w��n)���{�ϡ�Ȼ����Һ�ص���ϴ���^�V�ԫ@�Ú�������ǰ�(q��)�w��Ȼ���@Щǰ�(q��)�w�����c̼��䇻�ϣ���(j��ng)�^�ɲ��џ����õ���K�o����O����LiNFAO2��
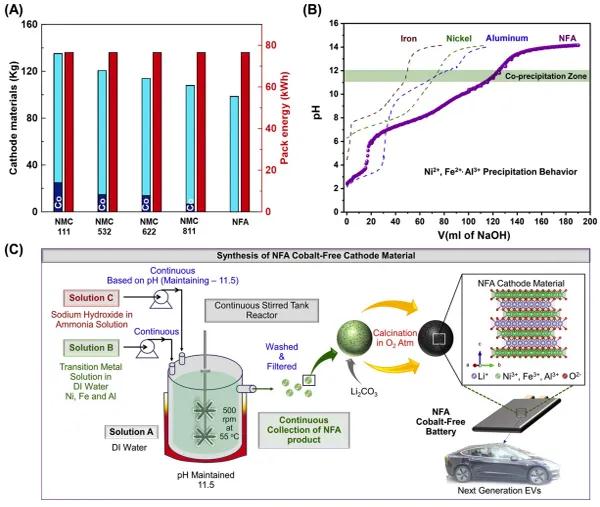
�D1.(A)�����ͬ260Ӣ��·���������O���ϼ���Ⓔ����ı��^��(B)Ni2+��Fe2+��Al3+�ȳ����О��NFA����Ԫ�ع������ı��^��(C)NFA�Ƃ��^��ʾ��D
����1.Ni2+��Fe2+��Fe3+��Al3+�l(f��)������r(sh��)��PHֵ����(du��)��(y��ng)��Ksp
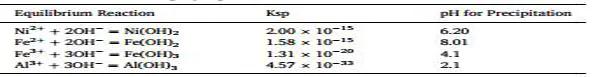
�Ò�������@�R(SEM)��(du��)�ϳɵ�ǰ�(q��)�w���Ӻ���K�����O��ĩ�M(j��n)����ò�������D2(A)����ʾ��SEM��������������ǰ�(q��)�w�����Σ�ǰ�(q��)�w��ƽ�������s��10 μm�������^�쵽����������ǰ�(q��)�w�ı���ʬF(xi��n)ɳõ���͈D��(�D2(B))���c���y(t��ng)��NCM��NCA��ơ�ǰ�(q��)�w���ӽ�(j��ng)̼����џ����SEM��D2(C)��2(D)��ʾ���џ���ķ��w��������ò�Ķ������ӽM�ɡ�����ͨ�^��(du��)���������џ���ˇ����(sh��)���M(j��n)һ����(y��u)���������õؿ��Ʋ��ϵ���ò�ͳɷ֡��D2(E)��ʾ��ϳ�ǰ�(q��)�w��400���A(y��)̎�����g�w����K���O��ĩ��X�侀����D��NFA��������ǰ�(q��)�w��XRD�D������ǰ�(q��)�w��α��β�ɷN����档Ȼ����NFA��������ǰ�(q��)�w������D�c��α-Ni(OH)2(ICDD-00-038-0715)��β-Ni(OH)2(ICDD-00-059-0462)XRD�D���ơ�Ȼ��ǰ�(q��)�w�cLi2CO3��ϣ����ڿ՚�����400���¼ӟ����џ���ǰ�(q��)�w�еĚ�������A(y��)̎��ǰ�(q��)�w��XRD�D������ǰ�(q��)�w�Ě���������ȫ�D(zhu��n)���γ��ˎr�}NFAO2�;��w�����A(y��)̎��֮�����O��ĩ������ĥ�������������џ����Դ��M(j��n)�Ƕ��NFAO2����Y(ji��)��(g��u)�γ���K���O��ĩLiNFAO2���D2(E)������D�f���@Щ�����γ��˺�������̼����s�|(zh��)�ĽY(ji��)�����^�ߵ����O��ĩ�����Կ���(006)��(102)��(108����(110)�ȷ���F(xi��n)���ѣ������γɵ��njӠ�α-NaFeO2�;��w�Y(ji��)��(g��u)��ͨ�^(003)��(104)�ķ及(qi��ng)�Ȍ�(du��)�ȁ��_���Ӡ����O�������(y��ng)�x�ӻ��ų̶ȣ�(003)��(104)�ķ及(qi��ng)�ȱȞ�1.7���f���(y��ng)�x�ӻ��ų̶Ȳ����@������(j��)���ޅ���(sh��)��ռλ�ʣ��_�����O���ϵijɷ֞�L(zh��ng)i1.0Ni0.85Fe0.052Al0.091O2�������F��NFA�wϵ�ĽM��Ԫ��֮һ����˹�����V���M(j��n)һ����(du��)�����M(j��n)�б�������˹�����V�W(xu��)�܉�_���F�������B(t��i)���r(ji��)�I�;��wλ�á����V�D���D2(F)���f��ǰ�(q��)�w����K���O���Ϸ�ĩ��혴��Եģ���(j��ng)�^400���A(y��)̎�������g���ϵĹ��V�^���(f��)�s�����Ǜ]�д����s�|(zh��)�V��ǰ�(q��)�w���^�����V�D�ֲ���������Ҫ����(g��)�M�ց팦(du��)��(y��ng)���r(ji��)�F���V�D���IJ��֡����⣬߀�^�쵽�������������r(ji��)�F��400���A(y��)̎�����V�����IJ����^��(ji��n)�Σ���Ҳ�������f���^���F�@ʾ���dz����Ĺ��V���Ɯy(c��)�c�o��������P(gu��n)��700�����џ����V�D׃���J�������dž�һ�����r(ji��)�F�M�֣��f���џ���С���ֶ��r(ji��)�F���������及(qi��ng)��׃?n��i)��f����ԭ���ֲ��V�����F��һ��(g��)�^խ�ĈD�V������(n��i)��׃����ˣ������Ɣ���џ��^���п����Ǵ��M(j��n)�Y(ji��)����Ҳ�����Ǵ��M(j��n)���������L(zh��ng)��
�D2.NFA��ĩ����ò�ͽY(ji��)���ȱ���
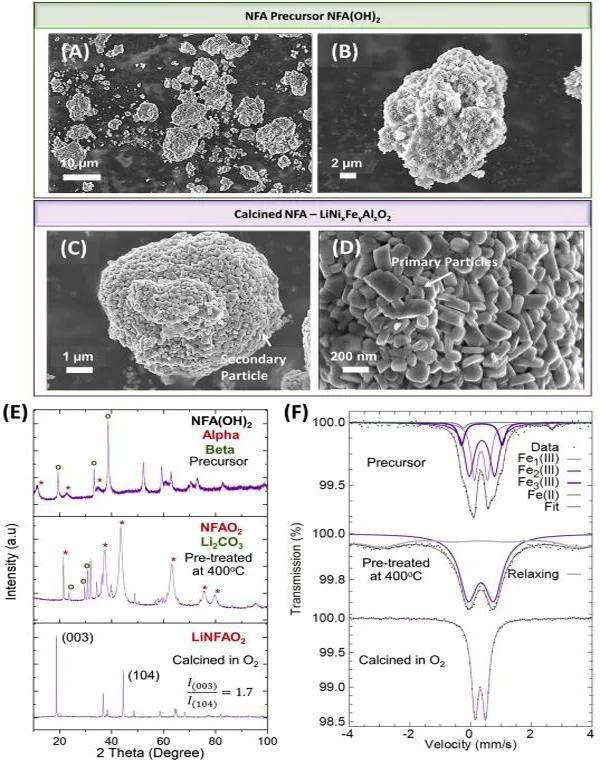
ͨ�^�M�bNFA���O���ϵ�2032�Ϳ�ʽ��늳y(c��)ѭ�h(hu��n)�����ͺ�������ԇ�(y��n)����3.0 V–4.5 V��3 V–4.3 V늉�����֮�g����0.1 mV/s–1 mV/s�Ē����M(j��n)��CV�y(c��)ԇ(�D3(A))����3 V–4.5 V�^��늉����ڷ�����(n��i)���քe��~3.75 V��4.0 V��~4.25 V���F(xi��n)���@��ÓǶ䇷塣ÓǶ������ھ����D(zhu��n)׃����i����߅��-1→��б(~3.75 V)����ii����б→��߅��-2(~4.0 V)�ͣ�iii����߅��-2→��߅��-3(~4.25 V)������߅��2����߅��3���D(zhu��n)׃�njӠ����O���������l(f��)��˥�p����ġ��ɂ�(g��)늉������е�CV�y(c��)ԇ�����������y(c��)�Ē����У�NFA���O�����^�õ�늻��W(xu��)�����ԡ��D3(B)��NFA���O�ڃɂ�(g��)늉������еĺ�������ԇ�(y��n)���@�N�o����O������0.1C���늱����£���3 V–4.5 V늉�������(n��i)�������_(d��)��190 mAh/g����3 V–4.3 V늉�������(n��i)������160 mAh/g��
�D3.NFA���O����늻��W(xu��)���ܵı���
�D4(A)��4(B)����(y��ng)��늳��迹�S늉���׃�����D4(C)�е�ʾ��D�f����R1��R2��R3���迹�����x���Y(ji��)��������R2�S��Ó䇶ȵ�������u�pС��ֱ��3.7V���ң��˺�ʹ�M(j��n)һ��Ó䇣������Ҳ�����ֲ�׃��R3���ֵ�S�������3.9 V��u�pС���˺��S��Ó䇳̶ȵļ�����׃�˽Y(ji��)��������NFA���O�������ӵ����늌�(d��o)�ʺ��x�ӔU(ku��)ɢ��׃�����������O-늽��|(zh��)����늺��D(zhu��n)�ơ����NFA�еij�ʼ�r(ji��)�B(t��i)��+3����������u�D(zhu��n)׃?y��u)�Ni4+��Ni3+/Ni4+�Ļ�σr(ji��)�B(t��i)����S���M(j��n)һ��Ó䇵����Ӷ����ӣ����γ�һ��(g��)��(d��o)늾W(w��ng)�j(lu��)���M(j��n)һ��Ó䇺�Ni3+��Ƚ��ͣ�Ni4+���ӣ���(d��o)늾W(w��ng)�j(lu��)��u���ѡ���ˣ����O늽�Һ�������R3���F(xi��n)���@һڅ��(sh��)���ĈD4A��BҲ�����^�쵽���S��SOC�����ӣ�R2�������ӡ��@�w���ڌӠ������Ó䇳�����ӌ�(d��o)��Լ������ӣ������(d��o)��R2�^С�����⣬��(d��ng)䇝���^�ߕr(sh��)��R3�^������ȫ䇻��A�Σ����ϵ���ӌ�(d��o)�������(du��)�^�͡�����EIS�Y(ji��)�������f������늺��D(zhu��n)�������һ��(g��)�����������أ��e���ڸ�䇝�ȅ^(q��)�g���D4(D)��SOC����x�ӔU(ku��)ɢϵ��(sh��)֮�g���P(gu��n)ϵ��40%SOCǰ�x�ӔU(ku��)ɢ���ʼ����½���Ȼ����u������70%SOC����70–100%SOC������(n��i)���U(ku��)ɢ�����_ʼ�����½����S�������څ��(sh��)�����^�ߵ�䇝�ȅ^(q��)�g���U(ku��)ɢϵ��(sh��)�^�ߡ��ڃɂ�(g��)늉������У����ϵ��x�ӔU(ku��)ɢ���S䇺�����׃��څ��(sh��)���ơ��x�����@Щ�����еĔU(ku��)ɢ����ͨ�^�g϶λ���λ�l(f��)�������g϶�͔U(ku��)ɢ�C(j��)���У��x�ӔU(ku��)ɢ����ͨ���SÓ䇳̶ȵ���������ͣ��ڿ�λ�͙C(j��)���У��x�ӔU(ku��)ɢ���S��Ó䇳̶ȵ����Ӷ����ӡ������õ��ĔU(ku��)ɢ����څ��(sh��)�D������NFA���O���F(xi��n)���g϶�͔U(ku��)ɢ�C(j��)�ơ����е�䇝�ȷ�����(n��i)����λ�C(j��)�ƿ����ǻ��S�ģ���Ó䇳̶��^�ߕr(sh��)����λ���������͔U(ku��)ɢϵ��(sh��)�����á��M����ˣ�NFA���O�ڃɂ�(g��)늉������еĔU(ku��)ɢϵ��(sh��)����3.5×10-12��2.5×10-12 cm2/s֮�g�����ýY(ji��)���c���y(t��ng)�Ӡ����O���ϵĽY(ji��)��һ�¡��@Щ�Y(ji��)�����������^�ߵ�ѭ�h(hu��n)�����£��x�ӔU(ku��)ɢ���ʿ�����һ��(g��)��Ҫ�ı����������ء�
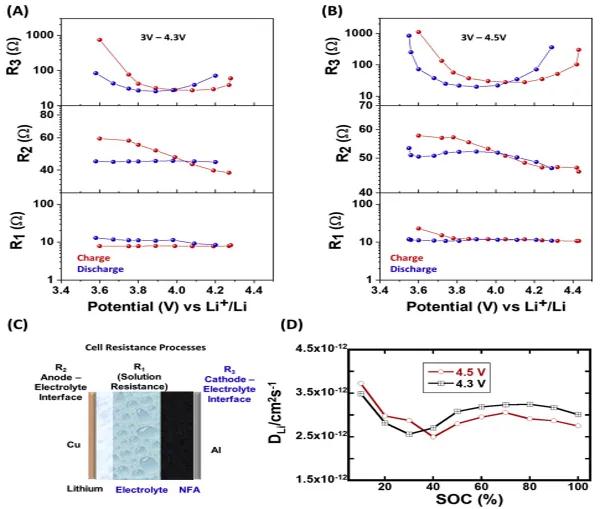
�D4.NFA���O���ϵ�늻��W(xu��)����
�D5(A)�ǃɂ�(g��)늉����ڷ�����(n��i)�ı������܈D����3 V–4.3 V��늉�������(n��i)��ԓ���ϱ��F(xi��n)�����õı������ܣ���0.1C�r(sh��)�����s��160 mAh/g��1C���ʕr(sh��)�������͞�s30%����3 V–4.5 V�^��늉�������(n��i)��ԓ������0.1C�r(sh��)�������s��190 mAh/g��1C���ʕr(sh��)�������������˼s40%�����⣬ͨ�^C/3�����ڃɂ�(g��)늉�������(n��i)�M(j��n)�г���ѭ�h(hu��n)�y(c��)ԇ��ѭ�h(hu��n)�����D��D5(B)��ʾ��3 V–4.5 V�^��늉������£����������s��180 mAh/g���S��ѭ�h(hu��n)�Δ�(sh��)�����ӣ���������u˥�p����(j��ng)�^100��ѭ�h(hu��n)�������s���ʼ������70%����(d��ng)늉����ڞ�3 V–4.3 V�r(sh��)�����O��������������Եͣ��s��150 mAh/g�����⣬���O�ڃɂ�(g��)늉������µĎ�(k��)��Ч�ʶ���99%���ҡ�3 V-4.3 V늉�����C/3������ѭ�h(hu��n)100�κ����������ʼs88%���@����?y��n)����^��늉�������(n��i)ѭ�h(hu��n)�r(sh��)����懲����аl(f��)������߅��-2����߅��-3����׃��(d��o)������˥�p��
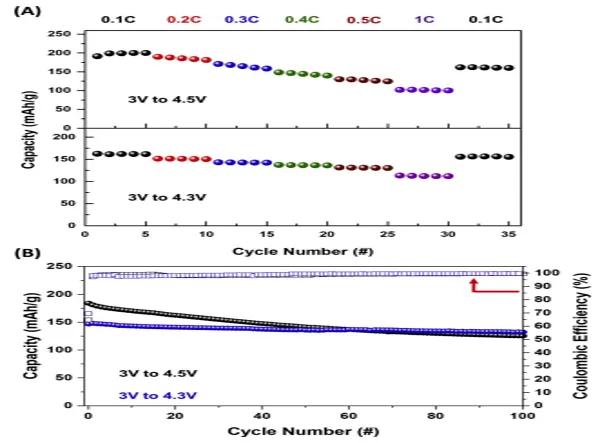
�D5.NFA���O���ϵ�늻��W(xu��)����
���Y(ji��)�cչ��
�C�������������״γɹ��ϳ���һ�N���͟o⒌Ӡ����O����NFA������(du��)���M(j��n)���˲��ϱ�����늻��W(xu��)���ܜy(c��)ԇ������CSTR�����������ɹ��ϳ�������NFA���Oǰ�(q��)�w����(j��ng)�M(j��n)һ��̎�����õ��˳ɷ֞�L(zh��ng)i1.0Ni0.85Fe0.052Al0.091O2�����O���ϡ�����X�侀�������˹�����V��(du��)�����M(j��n)���˱������l(f��)�F(xi��n)NFA���Ͼ��ЌӠ�Y(ji��)��(g��u)�����(y��ng)�x�ӻ�����١��ڲ�ͬ늉����ڗl���£����ö�N늻��W(xu��)�ֶΌ�(du��)NFA���O���ϵ�늻��W(xu��)�����M(j��n)���˱������Y(ji��)������ԓ���O��늻��W(xu��)�����c���y(t��ng)�ĺ��NCM��NCA���O���ơ����ʺ�ѭ�h(hu��n)���ܜy(c��)ԇ�Y(ji��)����������(d��ng)�����4.5 V�r(sh��)��ԓ������0.1C�ı��������_(d��)~190 mAh/g����늉�����3 V–4.3 V��ѭ�h(hu��n)100�κ����������ʞ�88%������3 V��4.4 V��늉��������M(j��n)����C/3��늻��W(xu��)�y(c��)ԇ����200�γ���ѭ�h(hu��n)����x��늳ر��F(xi��n)�����õ�ѭ�h(hu��n)���ܣ������s���ʼ������72%����ˣ��˷N���O����NFA�������I(l��ng)��һ���o���x��늳ء�
[�īI(xi��n)��Ϣ]
LiNixFeyAlzO2, a new cobalt-free layered cathode material for advanced Li-ion batteries(Journal of Power Sources.2020.DOI:org/10.1016/j.jpowsour.2020.228389)
ԭ��朽ӣ�https://www.sciencedirect.com/science/article/pii/S0378775320306935
(؟(z��)�ξ�������)
����(bi��o)����
늳�
�o����O����




��؟(z��)�������ăH�������߂�(g��)���^�c(di��n)���c�Ї�(gu��)늳�(li��n)�˟o�P(gu��n)����ԭ��(chu��ng)���Լ�����������ֺ̓�(n��i)��δ��(j��ng)���W(w��ng)�C��(sh��)����(du��)�����Լ�����ȫ�����߲��փ�(n��i)�ݡ����ֵ��挍(sh��)�ԡ������ԡ����r(sh��)�Ա�վ�����κα��C����Z��Ո(q��ng)�x�߃H����������Ո(q��ng)���кˌ�(sh��)���P(gu��n)��(n��i)�ݡ�
�����W(w��ng)ע�� ����Դ��XXX�����Ї�(gu��)늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c(di��n)�͌�(du��)���挍(sh��)��ؓ(f��)؟(z��)��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո(q��ng)?ji��n)�һ�܃?n��i)�M(j��n)�У��Ա��҂����r(sh��)̎����
QQ��503204601
�]�䣺cbcu@m.astra-soft.com
�����W(w��ng)ע�� ����Դ��XXX�����Ї�(gu��)늳�(li��n)�ˣ�������Ʒ�����D(zhu��n)�d������ý�w���D(zhu��n)�dĿ�����ڂ��f������Ϣ�������������W(w��ng)ٝͬ���^�c(di��n)�͌�(du��)���挍(sh��)��ؓ(f��)؟(z��)��
������Ʒ��(n��i)�ݡ����(qu��n)���������}��Ҫͬ���W(w��ng)(li��n)ϵ�ģ�Ո(q��ng)?ji��n)�һ�܃?n��i)�M(j��n)�У��Ա��҂����r(sh��)̎����
QQ��503204601
�]�䣺cbcu@m.astra-soft.com
����ϲ�g
-
ȫ�CҺ��늳صļ��g(sh��)�M(j��n)����Ч�÷���
2020-09-28 09:29
|
|
|

���}


���P(gu��n)��
-
ȫ�CҺ��늳صļ��g(sh��)�M(j��n)����Ч�÷���
2020-09-28 09:29
�����c(di��n)
-
2024�늳����Ј�(b��o)��
2024-05-24 18:59 -
С�����늳����죬�c���r(sh��)���������Y��˾��
2024-05-20 19:05 -
�y�ֶ���������@����I(y��)5������늳��(xi��ng)Ŀ�_��/���s!
2024-05-21 18:46 -
�ذ�������Դͻ���������ã��̑B(t��i)늳�ِ���������l(f��)
2024-05-28 18:18 -
��һ10GWh�(xi��ng)Ŀ�_�����̑B(t��i)늳ؾ��x�a(ch��n)�I(y��)��߀Ҫ��ã�
2024-05-11 19:17 -
���r(sh��)�����ȁ��ϡ��Є�(chu��ng)�º���ͬ��؛ε������������
2024-05-09 18:48
�gӭͶ��
(li��n)ϵ�ˣ���Ůʿ
Email��cbcu#m.astra-soft.com
�l(f��)���]���r(sh��)��@��Q#
�Ԓ��010-56284224
Email��cbcu#m.astra-soft.com
�l(f��)���]���r(sh��)��@��Q#
�Ԓ��010-56284224
�ھ�Ͷ��

©2017 ���(qu��n)���� �Gɫ�DžR��Դ���g(sh��)�о�Ժ �A����̩�Ƽ�������������˾ ���k Power by DedeCms
�r(ji��)ֵ�ɾ��ИI(y��)Ʒ�ƣ����\(ch��ng)�����ṩ���������YӍ
��ICP��2024061100̖(h��o)
�r(ji��)ֵ�ɾ��ИI(y��)Ʒ�ƣ����\(ch��ng)�����ṩ���������YӍ
��ICP��2024061100̖(h��o)
 �Ź���̖(h��o)
�Ź���̖(h��o)